Probably the most common line one can read in phylogenetic papers scratching at the surface of evolution is "species xxx is not monophyletic" or "supports species yyy as monophyletic". While appearing to be trivial, clade = monophylum, such statements are often naive, too often incomprehensive or, occasionally, just wrong. A clarification.
The concept
The phylogenetic species concept – and the typically considered synonymous cladistic species concept – defines a species in analogy to higher-level systematic units (generas, tribes, families, orders). Don't ask me who invented it. Willi Hennig, the father of cladistics (or, more accurately, the Kladistische Systematik), didn't bother about anything below the genus. Maybe because he actually read Darwin's famous book, The Origin of Species, in full (which I never did, I'm a visual guy and it only includes a single figure!) According to everyone I met who read through the entire Origin, Charles Darwin [Wikipedia], like other early evolutionary biologists and phylogeneticists (such as Alfred Wallace [Natl Geogr.] and Ernst Haeckel [DKU]), was too aware of inter-population, intra-specific and inter-specific variation to ponder there could be anything like an universally applicable species concept at all.
What makes the phylogenetic species concept appealing is its simplicity. Hennig provided a new, precise definition by distinguishing inclusive — monophyly — from exclusive common origin — paraphyly.
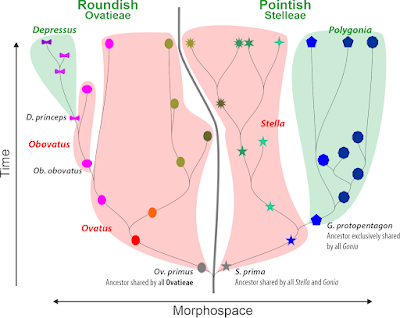 |
| Two reciprocally monophyletic tribes (Ovatieae, Stelleae) with morphologically defined genera, three of which are paraphyletic (red fields). Only Depressus, which evolved from an obovate precursor and Polygonia are monophyletic, since all known species (extant, modern-day, and extinct, existing in the past) can be traced back to an exclusive to them common ancestor, D. princeps and G. protopentagon, respectively. |
By assuming a phylogenetic tree, or knowing the 'true tree', we can hence easily test for monophyly. No matter at which level.
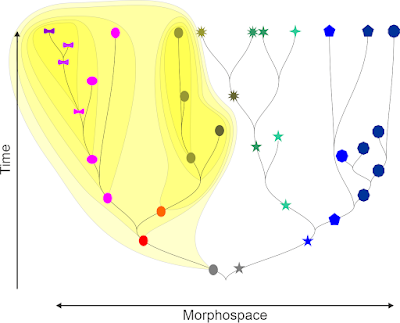 |
| All the yellow fields represent monophyla that can be definied within the Roundish lineage. They all are characterised by an inclusive common origin, including a common ancestor and all its descendants. While it is impossible to test directly for paraphyly, the test for monophyly is trivial. |
Accordingly, a monophyletic species is a collection of populations that share (go back to) a, exclusive to them, common ancestor.
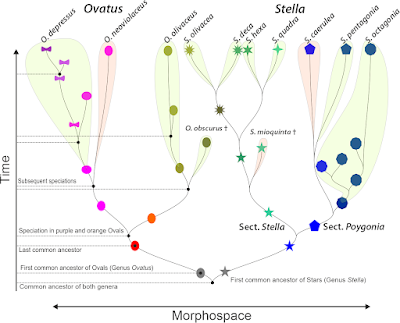 |
| Applying the Hennigian phylogenetic species concept to ovals and stars: the coloured fields represent the possible extention of the modern species into the past. Most of the deeper fossils must remain nameless, as they represent ancestors of more than one modern-day species, unless we drop all modern-day species and make both genera monotypic. Moon green: monophyletic species with discrete morphologies (qualifying for cladistic species), peach-coloured: species with ancestral morphologies, which probably would be misplaced even in a perfect total evidence tree: near-identical tips are always resolved as sisters as soon as there is any discriminative signal in the data to inform taxon bipartitions (here: purple and ovate; blue and pentagonal). |
Hennig didn't invent monophyly. Phylogenetic classifications that tried to identify natural, monophyletic groups from seemingly similar but not related, hence invalid, polyphyletic groups surfaced similtaneously with the general acceptance of the concept of evolution and phylogenetic trees (Who published the first phylogenetic tree?). From the late 19th century onwards, more than 50 years before Hennig separated ‘monophyly’ s.str. and ‘paraphyly’, biological taxa (series, sections, genera and higher) were increasingly defined by having a common origin, ‘monophyly’ in a general sense, the hypothesis that they go back to a shared ancestor from which they evolved.
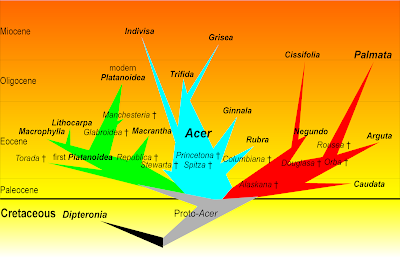
For instance, Schwarz (1936) explicitly states that Camus' classification of oaks (1934; 1936–1954) is Lamarckian-phenetic, hence, “unnatürlich” [un-natural] since being based on superficial similarity, while not considering the “phylogenetischen Beziehungen” (phylogenetic relationships). Pojárkova (1933) included an explicit evolutionary (Darwinian) tree in her monography of maples (genus Acer), where some of the series and sections were placed as ancestors (precursors) of others or as sister lineages. It inspired me to come up with this figure in my Ph.D. thesis (Grimm 2003, Fig. 4-26) to summarise my molecular-phylogenetic results on the background of an earlier morphology-cladistic study (Wolfe & Tanai 1987).
Noting the semantic conflicts, Ashlock (1971) proposed to use a new term for Hennig's monophyly, ‘holophyly’, to discern it from the general meaning of monophyly: common origin (irrespective of inclusive or exclusive). A paraphyletic taxon is monophyletic taxon that is not holophyletic. Because this is a very useful distinction when talking about "monophyletic species", I will follow Ashlock in this post (something not really possible in a phylogenetic paper, we only went through with it once: Bomfleur et al. 2017)
- Holophyletic = inclusive common origin in a strict Hennigian sense: a common ancestor and all its descendants.
- Paraphyletic = exclusive common origin in a strict Hennigian sense: a common ancestor and some of its descendants.
- Monophyletic = either holophyletic or paraphyletic.
Let's call this the holophyletic (Hennigian phylogenetic) species concept:
A natural species is a holophyletic group of populations.
Thus, it includes all populations of an organism that can be traced back to the same, and exclusive to them, common ancestor.
To get not entirely lost in philosophy, we will acknowledge that this common ancestor may have been a single population or already a group of inter-connected populations—a network of populations where inbreeding (gene flow within the population) has not yet outcompeted outbreeding (gene flow between populations). We will also acknoledge that a population can be anything between a single stand and a patchwork of scattered individuals; and that there are in principle two kinds of ancestors, homogenous and heterogenous ones. For instance, the ancestral species AB and DE in the following graphic have multiple ancestral populations, and species S* is of hybridogenous origin subsequently intrograding into one parental lineage and taking it over: the extant population have two, exclusive to them, common ancestor: the original hybrid (A × B) and the ancestor of the lineage (T), an offspring of B, they took over.
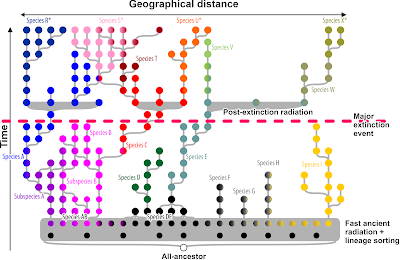 |
| Populations over time forming holophyletic species. Surviving (extant) ones indicated by asterisks. The filling represents the genotype(s) of each population; gradients indicate mixed/incompletely sorted gene pools. The dominant gene pool typically defines the phenotype (ring colour) |
Note that from a total evidence perspective all precursors (A, C, E, T, W) are per definition paraphyletic. But at a certain point in time, they were holophyletic. For instance the post-crises (after the major extinction event) re-radiating A, C and E populations all go back to a single survivor each. Only with the evolution of their offspring (R* and U*, hybridogenous S*, extinct T), they become paraphyletic. Pre-crises species A, B, and C are also reciprocally holophyletic as they can all be traced back to a single or two populations that are not shared with their siblings.
If the populations of pre-crises species A and C would be genetically distinct, we would have little problems to place also the populations of species B in a distinct clade.
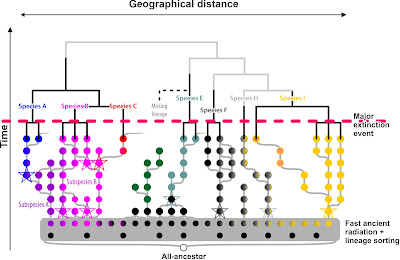 |
| Cladogram depicting the phylogenetic relationships between pre-crisis populations. All species clades represent holophyla, their respective, exclusive to them, first common ancestor(s) highlighted by stars. |
Given our (molecular) data well reflects the evolutionary history (morphology is generally useless for tree inference at this level), each holophyletic species will form a clade in an inferred tree. Which is the basis for the cladistic species concept.
Identifying cladistic species is trivial; strikingly simplistic (hence, much loved, we all like it simple, don't we?).
If my individuals form a clade in the inferred tree, they must be holophyletic.
Note that while all our (pre-crisis) holophyletic species have a good chance to from clades, their inter-species relationships (the grey branching patterns) cannot reflect the reality, because we started with a fast ancient radiation (the typical real-world situation for any genus that is not a relict but still expanding today).
Imagine, we have a time machine and can go back to sample the populations of our lineage at different time scales:
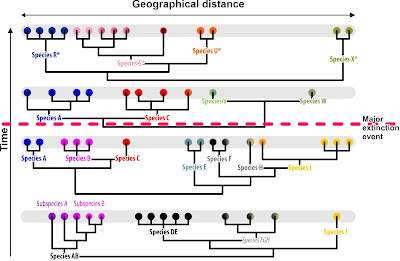 |
| Assuming we'd have a time-machine. Cladograms depicting the likely inferred phylogenetic relationships for co-eval populations and the subsequently defined cladistic species |
The species clades in our thought-experiment above are holophyletic at their time (see above). One practical shortcoming of the cladistic species concept is obvious: we only can select the clades to put names on (the filling of each population dot shows its genetic affinity) because we start from a prior (morphological, the ring colour) classification scheme. Without this essentially ‘phenetic’ framework, we'd be lost, as the deeper clades (where lack of lineage sorting obscures the signal) may refer to holophyla but some are reconstruction/data branching artefacts. On its own, the cladistic species concept is impossible to apply unless we have additional means to assess the evolutionary quality of a clade (we'll come to that later). Thus, we only use it to test a prior phenotypic, typically phenetic, scheme. For instance, the molecular phylogenetic tree tells us that species F and H, morphologically highly similar or indistinguishable are not part of the same clade, hence, they must be (pseudo-)cryptic species. Theoretically, inferring clade to identify holophyly could work. Nearly.
With morphology in mind as guideline tested via our molecular clades, we would end up with one paraphyletic species, though, at the time slice just after the initial fast radiation: species FGH. The geographically triggered genetic closeness of F, G, and H so close to the primary radiation would result in resolving them as a sister clade to the genetically homogenous (coherent), non-evolved (primitive) but at this time distinct from any other lineage, species DE (a sure clade). Being morphologically similar (grey ring), we would have no reason to distinguish them as the (pseudo-)cryptic species they are.
Blessed are cladistics that we don't have time-machines, right? But, today, depending with genus we look at, we may be still close to the intitial radiation (the lowest cladogram in the graph), at a time extinction and isolation has sorted things out, finely so, or at the time where re-radiation led to secondary conflict.
When invoking the cladistic species concept (but see e.g. the PhyloCode initiative, ending up with circumnavigating the species naming question), we blindly accept that inferred clades are synoymous with Hennig's monophyly (= holophyly):
A clade in a rooted tree is a necessary and sufficient criterium for holophyly.
[see Wikipedia, in case you are unfamiliar with necessity and sufficiency in logic and mathematics]
Funningly, while often invoked ("we tested the monophyly of species"), it's rarely used consequentially — we seem to be content in noticing "non-monophyly" but rarely come up with re-describing species. Even so the diagnosis of a cladistically defined species or higher taxon would be pretty easy to do (one could follow the PhyloCode guidelines), e.g. for our first example of ovals and stars:
- Ovatieae — any genus that is part of the corresponding clade.
- Ovatus — any species that is part of the corresponding clade
- Paraovatus — any species that is part of the corresponing clade
- P. depressus — any individual/ population of the clade containing the precursor-taxon formerly known as D. princeps.
- ...and so on...
- Stellatieae — any species/individual that is part of the corresponding sister clade.
So, why are we talking about and testing a species' "monophyly" but still add descriptive features? Such as:
- Ovatieae — often oval-shaped, roundish.
- Ovatus — orange or greenish, ovate and oval-shaped.
- Paraovatus — obovate or donut-shaped, pinkish
- P. depressus — always donut-shaped
- ...
- Stellatieae — pointed, star-shaped or hexagonal
Facing reality
To apply the cladistic species concepts, we only have to assure (or be sure of):
- Speciation is a dichotomous process that can be modelled via a 1-dimensional rooted tree (the one dimension being time). Note that it can involve simple reticulation as in our theoretical example, e.g. species S* (the PhyloCode calls them “clades that are partially overlapping”) or pre-crisis species B but keep in mind the problem posed by fast ancient radiations.
- The biological species concept (proposed by Ernst Mayr, who hated the guts of Hennig, see e.g. this late piece: Mayr & Bock 2002) applies as well: once a species evolved, it cannot produce (a lot) fertile offspring with individuals outside the species. In other words, there's no or only very little hybridisation, partial introgression, allopolyploidisation but more importantly: no incomplete lineage sorting and little intra-species, inter-population variation.
- Species manifest via cladogenesis, i.e. each speciation event is accompanied by the accumulation of lineage-specific/ -shared characteristics (ideally derived, synapomorphies fide Hennig, or homoiologies). [see also this Geneal. World Phylogenet. Networks post]
- We have methods to infer the 'true tree' from the data available. And ways to identify false positives: (well-supported) clades that are data-/method-artefacts (like species FGH in our thought experiment).
Quite a bunch of implicit assumptions, we rarely ponder when writing "our data reject/ support the monophyly of xxx".
I worked over 20 years at the coalface of evolution (sometimes literally, one finds nice plant fossils in lignite-coal mines): the transition zone between populations, species, and genera. And we didn't restrict ourselves to the now, but kept a close eye on the historical past, the fossil record of our study objects (when available). We used species and other taxa as real (existing) spatio-temporal biological units.
My Ph.D. thesis (Grimm 2003) was about the “Mode and Speed of Intrageneric Evolution” in maples (genus Acer) and beeches (genus Fagus), two common and pretty ordinary northern hemispheric tree genera that, at the time, were oddly understudied by systematic botanists and molecular phylogeneticists.
I realised very quickly and was frequently reminded (see papers collected on my GoogleScholar profile and literature cited therein, most of the recent ones are open access paid by Swedish and Austrian tax-payers, thank you!):
- Speciation is not a dichotomous process. But a gradual, often messy one. Or polytomous: a widespread, already genetically heterogenous species breaks up not in two but many species precursors because of area fragmentation (e.g. changing climate). Or reticulate, and possibly involving multiple layers of "overlapping clades" (or, as we prefer to call them: "evolutionary lineages")
- Plants, in particular, would have enjoyed the Origin as a good read but would have laugh their leaves off when reading Mayr. As long as they have a similar karyotype, and come close and cosy, they don't give a shit about species boundaries. Most of them see little point in telling a bee or alike: please don't carry my pollen to that other species. And some even bother so little, they just disperse it via the wind. And if the karyotypes are too different, there's always the option of allopolyploidisation to get through a nasty bottleneck situation or going bold and invasive fueled by hybrid vigor. This doesn't they don't establish coherent species at all, e.g. in any mixed oak stand it's possible to unambiguously identify different species, even though they belong to the same evolutionary lineage and mix, like the European widespread, often sympatric white oaks Q. robur and Q. petraea (two essential reads: Neophytou et al. 2010, Lepoittevin et al. 2015). But the mixing is too limited to fuse the species.
- There's hardly any cladogenesis associated with speciation. Cladogenesis is the end-product of sorting following speciation and isolation events (can take millions of years in plants) or evolutionary bottlenecks:
- Specific traits are gradually accumulated in a lineage.
- If beneficial they tend to quickly dispers across the phylogenetic neighbourhood. Convergences, random similarities, and general homoplasy are bad for tree inferences, but parallelisms (the tendency to evolve or express a trait within a group of relatives) and positive selection are worse.
- There are no synapomorphies below the genus level, at best we have homoiologies and often its just specific suites of characters.
- Morphologies in general but genes as well suffer from incomplete lineage sorting.
- Phenotypes are not generally congruent with genotypes. Some traits are not the product of the evolutionary history of a genus, but their current ecological niche (keyword: epigenetics)
- When going after the species trees, visualising the rate of change is crucial: cladograms are pointless graphs, phylograms obligatory but may still be incomprehensive. To illustrate the evolutionary history of a genus you need at least an evolutionary tree (like Pojarkova's) (or "cactus" or "coral") depicting explicit ancestor-descendant relationships. If not a species network.
- There's no perfect data. For any data set one can pull together applies: a (hopefully) high number of propects comes with (hopefully) few but usually severe, sometimes inevitable pitfalls. Thus, will ultimately lead to aspect-wise wrong topologies. False positives, clades that are definitely not holophyletic but data-, model- or method-based artefacts. And the most critical of them, will have high or even unambiguous support. Never blindly trust an inference, always explore your data. [For examples and further references see tag EDA, short for ‘exploratory data analysis’ (e.g. Morrison 2010), @ dormant Geneal. World Phyl. Networks.]
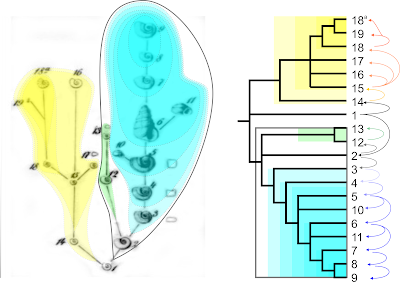 |
| Left: One of the first explicit phylogenetic trees ever published (Hilgendorf, 1866). Right: how it would have been depicted in a typical contemporary molecular systematic study. Coloured fields represent the holophyla. 150 years ago, the aim was to reconstruct evolution, now we are content with depicting (often data-wise trivial) sister relationships. |
Consequently, it's usually impossible to simply infer a (single) tree that matches the 'true tree', not to mention the 'true network'. Especially in the case of genera, and even if one can tap into phylogenomic data sets. No matter how high the support (common thresholds still being bootstrap support ≥70, Bayesian posterior probability ~ 1.00), an inferred clade is neither a sufficient nor a necessary criterion for holophyly. Especially not in the case of species.
In cladistics for a group of organisms, monophyly is the condition of being a clade[sic!]—that is, a group of taxa composed only of a common ancestor (or more precisely an ancestral population) and all of its lineal descendants [that should include extinct, not mentioned in e.g. the PhyloCode, and extant; hybrids, introgrades, allopolyploids make any holophyletic group para- or polyphyletic, as they have two (or more) ancestors, shared with different groups]. Monophyletic groups are typically characterised by shared derived characteristics (synapomorphies), which distinguish organisms in the clade from other organisms [fun-fact: one of the genetically most obvious angiosperm clades, the Magnoliids, have "features as in 'Early Angiosperms', i.e. the so-called para-/polyphyletic 'ANITA grade'; check out the great APG IV Poster]. An equivalent term is holophyly. — Wikipedia: Monophyly
I also learnt another important lesson (especially when it comes to publishing in single-blind peer-reviewed journals, and your peers being inevitably cladists):
If you want to get a nice, fully resolved species tree, never ever generate molecular data for more than a single individual per species (or genus, when doing intra-family phylogenies).
But also:
If you want to get a close-to-reality-as-possible species network (which may be a tree but not necessarily the easily inferred one), always include as many as possible individuals per species. And ready the weapons for battling the Mighty Beasts of the Forest of Reviews.

Reasons for “non-monophyletic” species
I recently got to review a pretty nice paper for a neat little (low-impact but green open access) journal with very interesting data. But (to the authors' dismay, I got the feeling) not leading to one of those trivial, fully resolved species trees. The authors made the same errors, I never stopped (and enjoyed!) doing:
- They looked at a biparentally inherited gene region. It's much easier to get trivial, albeit incomprehensive or severly misleading (Are complete plastome trees always better?), trees when the genomic information is only passed on via mothers, mitochondrial and chloroplast genomes in many organisms, or fathers (e.g. Y-chromosome in mammals).
- They included at least two individuals per species (see above).
- They looked at genus, very few people bothered to sequence so far. Especially in the age of next-generation sequencing and Big Data, one reason a genus may be sparsely covered in gene banks and phylogenetic literature is that everyone who looked into it, saw it's a pit of wriggling mulit-headed hydras, and quickly dropped the project. It's publish-or-perish not Viel Feind, viel Ehr' [the more enemies, the greater the glory] in science.
Hence, like me, they ended up with the often invoked "non-monophyletic" species, i.e. individuals not coming up as part of the same, well-supported clade in the inferred (typically outgroup-rooted) tree.
The failure to place individuals of the same species in the same clade – “non-monophyly” in molecular-phylogenetic literature – may however have very different reasons:
- The species is holophyletic but the used molecular data hasn’t been sorted during speciation, i.e. is (partly) at odds with the phylogeny, the evolutionary history of a genus. This includes (overlooked) paralogy, homeology and pseudogeny topological effects. And many aspects of incongruent genealogies.
- The species is holophyletic but the used molecular data (or used model) lacks the capacity to resolve this. E.g., when your genus is very young, as are its constituent species, you wouldn't expect sorting and inbreeding having already homogenised each species' gene pool to a degree that you can discern them genetically and phylogenetically via a tree. If the level of divergence is very low, probabilistic methods easily get lost in the flat tree space: they cannot resolve near-identical tips. The clade will have minuscule roots and little (bootstrap, BS) support. Note: Bayesian posterior probabilities (PP) may still be high, pending how trapped the Bayesian chains got in suboptima: Mind any clade with high PP and low BS! The mere lack of support cannot reject holophyly (or monophyly in general). Parsimony and distance tree inferences will always fall in the trap of long-branch attraction (LBA), probabilistic methods have a 1:1 chance to get it right or wrong in the ‘Felsenstein Zone’ — no outgroup is always better than using a too distant one! Keep in mind that no standard tree inference can handle explicit ancestor-descendant relationships. With low-divergent data, (parsimony) haplotype networks are currently without alternative.
- The species is paraphyletic, and one or more species evolved from this species. In molecular trees, paraphyly can lead to moderately supported clades with short root branches. One reason for paraphyletic species is "budding speciation", the isolation of a holophyletic offspring from a larger species. Given time budding speciation will lead to "reciprocal monophyly", sister clades, in molecular trees: either the remaining populations will keep on homogenising their gene pools (leading to holophyly) or having large population sizes, they are not affected by genetic drift to the same degree as the off-shot (‘short-branch culling’; inverse LBA). Well-resolved paraphyletic grades are rare in inferences, as they require the populations of the paraphyletic species (or taxon in general) to be genetically isolated from each other, i.e. being composed of cryptic or pseudo-cryptic species. More often, they are expressed as poor resolution: several adjacent, short, poorly supported branches. While considered "not valid" in cladistics, identifying and classifying paraphyletic species (or groups) is paramount for the understanding of intrageneric evolution because they inform us about ancestral morphologies, extant species that are still relative close to the common ancestor of a species group, and leftovers of earlier radiations before the crown groups evolved and took over most of the genus. Their (non-exclusive) common ancestors, be it the first or last common ancestor, will typically be characterised by a profoundly primitive suite of characters.
- The species is periphyletic or epiphyletic (Wheeler, 2013, Cladistics 40: 447–451). Nice concepts for monophyletic groups that are not holophyletic. Not sure ever used (e.g. not mentioned in the PhyloCode; see also D. Morrison's post Monophyletic groups in phylogenetic networks). As soon as there was hybridisation, introgression, allopolyploidisation or any kind of lateral/ horizontal gene flow involved in the evolutionary history of a genus and the formation of species, any inferred species tree (including multi-species coalescents) must be incomprehensive. Note that the resultant monophyletic but definitely not holophyletic species will usually produce high-supported clades when using data passed on via only one parent such as chloroplast and mitochondrial genomes or the Y-chromosome. Using biparentally inherited nuclear genomes, any form of reticulation is expressed as topological ambiguity: in trees typically by long/ prominent but poorly supported (bootstrap!) branches. PS For plants, the rule-of-thumb is: nuclear genealogy = phylogeny, inter-species relationships (species network); plastid genealogy = geography, species' migration history.
- The species is polyphyletic/insubstantial. There are quite some bulk taxa erected for individuals not fitting into the well-circumscribed species of a group. Only for relatively few genera, deep data are available to assess the morphological coherence of described species. The floras of the world are still full of taxonomic bias, species described based on exotic herbarium sheets by botanical experts who never set food into nature. Like de Candolle, who had agents browsing through the wilderness sending material to Paris, much of which is still unpacked (Denk et al. 2012). Tenore described pan-Italian endemites for the glory of the finally unified Italian state. Nationalist naturalists like him were common around 1900. A side effect of the Cultural Evolution in P. R. China was to rid the Flora of China of imperalist species (described by Europeans or, worse, Japanese) by inventing Chinese endemites; a sport that recently took up speed again (see the many published P. R. China-based single-/few-plastome studies focussing on microspecies).
- An individual has been mislabelled or the material was contaminated. Not uncommon reasons for ‘non-monophyly’ in plant molecular studies in the 90s/early zeroes, or more recent barcoding studies (one extreme example [comment to a PLoS ONE paper] I accidently crossed during one of my gene bank harvests).
- Individuals include morphologically inconspicuous F1-hybrids. In groups with many assumed, morphologically postulated hybrids, i.e. groups with poorly described species boundaries, mislabelled individuals are inevitable, even when one carefully revises the used collection material. Next-generation approaches rely heavily on cultivated material from arboreta, nurseries, and botanical gardens. Species who would never come into contact in the wild are kept close, and cultivation hybridisation becomes possible. Note that too many molecular-phylogenetic studies still don't involve any taxonomic expert at all, the researchers blindly rely on the tags put on the material they processed (or harvested from gene banks).
Mislabels and cryptic F1-hybrids can only be filtered out by relying on large individual and species samples. For instance, in our studies of west-Eurasian maples (Acer sect. Acer) and oaks (Quercus) but also in plane trees (Platanus) and related studies, one can find:
- Morphologically intermediate individuals showing genetic signals from both assumed parents, e.g. an individual growing in the botanical garden of Naples which looks like a Quercus trojana but has corky branches as only found in Q. suber-crenata and shows the specific nrDNA variants of both species (included in Denk & Grimm 2010). From southern France, we collected maples morphologically intermediate between Acer monspessulanum and A. opalus, showing both parents' highly diagnostic ITS variants (and even recombinant copies; Grimm et al. 2007, fig. 1 and appendix B).
- Individuals of morphotypes purported to represent hybrids that are genetically homogenous (pseudo-hybrids). Hybrids/ intermediates only occurring with one of the assumed parents, or showing stable morphologies even in contact with their other parent. For instance, there's no genetic evidence so far for a hybrid origin of Quercus afares or Fagus x moesica.
- Known hybrids that are genetically (seemingly?!) homogenous because of frequent backcrossing with one parent: the 'London Plane' (Platanus x hispanica/ acerifolia, a cultivated hybrid between the eastern North American P. occidentalis and the eastern Mediterranean P. orientalis may or may not show nrDNA signatures of their North American ancestors.
- Cryptic hybrids, morphologically inconspicious individuals/ populations where the genetics just don't match up.
- Variants/ subspecies that are actually of hybrid origin, even though taxonomists never thought of it, such as Platanus palmeri (Grimm & Denk 2010) according to Flora of North America the southwestern subspecies of P. occidentalis)
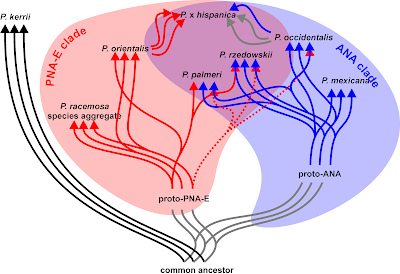 |
| Inheritance of three nuclear non-coding markers — two multi-copy, showing intra-individual polymorphism, one single-copy showing heterozygosity – in Platanus (plane trees; Denk & Grimm 2010) indicating old – formation of SE U.S./NE Mexican P. palmeri and N/C Mexican P. rzedowskii; ancient gene flow between PNA-E and ANA clades – and recovering known recent hybridisations (‘London Plane’ P. × hispanica = 17th century hybrid of P. occidentalis and P. orientalis) |
Should we bother about holophyletic species?
For most research, the sampling is more important than the names. But when it comes to assessing the validity of names pinned on organisms, we do want names that have a defined evolutionary quality, and including the species level. When we talk about biodiversity in higher organisms (plants and animals), what we mean is number of species. The prime philosophical questions are:
At which point turns lateral gene flow, hybridisation or introgression a holophyletic species into a non-holophyetic (epiphyletic, periphyletic) one?
To which degree can I discern holophyly of a species at all, in contrast to other types of monophyly (paraphyly, epiphyly, etc...)?
Obvious is:
Testing holophyly of species solely by inferring molecular trees is pretty pointless. Especially when I only have gene sample at hand inherited from one parent only (chloroplast and mitochondrial genome data in [most] plants, mitochondrial data and sex chromosomes in animals).
Equally pointless is trying to apply a strict Hennigan phylogenetic or cladistic species concept at all to replace the current morphology-based ones. The closer we get to the surface of evolution, there will be many cases in which it will be impossible to discern holophyly from other kinds of monophyly. The broader the sample, the higher the chance of alternative branching patterns (topological uncertainty), competing clades.
Well-inferred clades are a near-sufficient criterion for monophyly in general, but not for holophyly in particular.
Save for cladistic reviewers, I never found any compelling reason to drop a species, just because it turns out to be not holophyletic. Monophyletic will be enough. Also phyly changes over time: Paraphyla can become holophyla, holophyla epiphyla or paraphyla, and so on.
Last but not least:
Linnéan binominals based solely on inferred clades are superflous and useless.
If I define taxomic units like species exclusively by placing them in an inferred tree, I can simply do that. Why bother to name the clade? Sampling bias is a long known issue in phylogenetics: I anyway have to reinfer the tree with every new and not (near-)identical sample (or deeply nested in a crown-group) to make sure it doesn't change the tree, hence, my clades. So, everyone can just look up in the currently valid tree which tips form a clade. The tree also defines the phylogenetic distance between every tip, in case I want to quantify biodiversity in the tree. Cladistic classification doesn't need names, only well-inferred trees based on a firm data fundament.
They are useless because I cannot expect a biodiversity curator (forester, census field biologist, field ecologist) to sequence every individual for inclusion in a global, sanctionised tree, only to tell some politicians how many species (each of which is a unique genetic resource) will go extinct when they burn down this patch of nature to make place for, e.g., biofuels. And to search for and come up with genetic-identification markers (‘barcodes’), I need a stable-as-possible – independent of sampling and inference models – and empirically applicable species concept to start with.
Don't stop with holophyly!
Molecular genetics can qualify (and quantify, different story) the evolutionary meaning and correct/ refine the empirical species (cf. the many works by Mallet, some examples provided in the citation list) that are distinguished and generally accepted (e.g. listed in the various floras and faunas). Natural species that, eventually, can be genetically confirmed using broadly sample data sets and represent useful taxonomic/ biological units. Like, for instance, our Acer orthocampestre [EuroMed/Kew Global Checklist], the ‘Actual’ Field Maple, a cryptic and genetically ancestral maple species of the Caucasus, morphologically indistinguishable from the ordinary (and now holophyletic again, thanks to the removal of the Colchic populations) Field Maple, A. campestre found across the rest of western Eurasia (Grimm et al., 2014). By recognising its genetic uniqueness and giving it a name we highligh its importance as a distinct genetic resource and stress that loosing the Caucasian Field Maple populations means loosing a unique species. Identification of (pseudo-)cryptic species is straightforward based on genetic differentiation and having a phylogenetic background for the entire lineage.
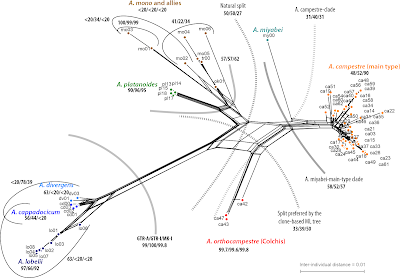 |
| Defining good species. The genetic diversity patterns are illustrated by a neighbour-net based on inter-individual 'phylogenetic Bray-Curtis' distances (Grimm & Denk 2014, fig. 3). Putative monophyletic/ holophyletic species are fairly obvious (labelled) as well as the genetic uniqueness of the new cryptic species A. orthocampestre collecting the Colchic individuals of A. campestre (s.l.) The brackets and splits with numbers refer to probabilistic (tree) branch support of topological alternatives (competing, partially incompatible clades and subtrees) using three different maximum likelihood implementations (GTR-A, GTR-I, and MK-I, cf. Potts et al. 2014) |
Another example from our own research how to fuse genetics into taxonomy and the description of species can be found in Liede-Schumann et al. (2019), where we simply included the (highly diagnostic) genetic sequence features into the new and emended species diagnoses.
But there's more to explore than refine taxonomic concepts and get better circumscribed species. Trying to reconstruct the actual evolutionary history of a genus and its species.
Towards this end:
- A clade in a nuclear-based (biparentally inherited gene sample) is a good indication for monophyly in general. As is a distinct neighbourhood in a neighbour-net splits graph (see maple example above). Note that monophyly in general but especially holophyly (original or stabilised post-speciation) typically expresses itself in highly coherent neighbourhoods when using neighbour-nets (Bryant & Moulton 2004), which I stronly recommend at the intra-generic level (fast and 2-dimensional).
- A well-hung clade (subtree in an unrooted tree) is a near-sufficient criterion for holophyly. A well-hung clade has not only appreciable, competetive branch support (any alternative not or weakly supported) but a prominent root branch (use the phylogram!) associated with substantial character support (check out RAxML's and IQ-Tree's new functions). If you sneak-a-peak into the alignment, and struggle hard to find any clade-unique alignment patterns even in the most variable gene bits, your clade may just be a reconstruction artefact.
- High clade/ subtree/ neighbourhood coherence in any sampled gene region, is a quasi-sufficient criterion for holophyly: intra-group variation < inter-group divergence. The higher the difference, the better.
- A well-explored species network that joins as many lines of evidence as possible is a necessary criterion for any kind of monophyly. It allows to discern between holophyly, epiphyly, periphyly, paraphyly etc. Or to simply identify the variously shared common origins. [PS: a tree is after all only a network without reticulations.]
An example for all sorts of monophyletic species: the beech
The beeches give a brilliant example. According Jiang et al. (2021) and implicitly following the cladistic species concept, they found that all modern-day species are “monophyletic”, i.e. holophyletic. Their up to four individuals (each represented by two phased, reconstructed “haplotypes”) per species formed high-supported clades in their combined tree (the multi-species coalescent, however, was astonishingly cloudy but didn't trigger to question the found species holophyly). But if one looks into the data behind their trees (combined and coalescent; Cardoni et al. 2021, detailed in sections 4.5f in Data S1) and adds further lines of evidence, it becomes clear that although they are all monophyletic, i.e. share a relatively recent common origin, only a few are holophyletic.
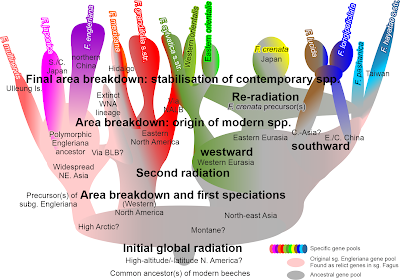 |
| A species coral for genus Fagus, the beech (Fig. S21 in Data S1 of Cardoni et al., 2021; for data see Grimm, 2021). |
The western Eurasian beeches are holophyletic in origin. We can even put a name to their first (FCA) common ancestor(s): the westward migrating †F. castaneifolia (cf. Denk & Grimm 2009). But the roots of the extant European F. sylvatica, the European Common Beech are epiphyletic. It shares an (inclusive) common origin with F. orientalis, the beeches (!) of the Orient (SE Bulgaria, NE Greece to N Iran), their last common ancestor(s) (LCA) are known as †F. haidingeri. F. orientalis as currently circumscribed is paraphyletic (obviously so, it's not even morphologically homogenous: Denk 1999) and includes at least two distinct species: the possibly periphyletic or paraphyletic to F. sylvatica Western orientalis and the (nowadays) holophyletic Eastern orientalis, a local and early diverged isolate of the once widespread F. haidingeri species complex. The Common Beech (F. sylvatica) is epiphyletic because it also inherited genetic material from a North American lineage of beeches that crossed into Europe via the North Atlantic Land Bridge († F. gussonii ?). The Eurasian ancestral species (†F. castaneifolia → †F. haidingeri) were all paraphyletic: they include(d) the common ancestor(s) of the sister species F. sylvatica + western F. orientalis as well as the precursor(s) of the eastern F. orientalis, and possibly even the FCA of the Japanese F. crenata, the closest living relative of the western Eurasian spp. [For the whole story see: The challenging and puzzling ordinary beech – a (hi)story.]
With the western Eurasian species having evolved from the same stock than F. crenata, Jiang et al.'s unambiguously supported “East Asian clade” must hence be paraphyletic. In fact, it is a reconstruction artefact (A fully resolved, perfectly misleading species tree), a fine example for a false positive (and the pitfalls of cladistic classifications). It also prooves that highly supported clades in combined trees reflect monophyly much better than holophyly. Fagus crenata is a today (near-)holophyletic species with a very dynamic past. Its precursors likely shifted back-and-forth between paraphyly-holophyly (in phases of range shrinking and isolation) and epiphyly (in phases of range expansion and introgression). Fagus longipetiolata might share one inclusive common origin with F. hayatae s.l. The link got lost in Jiang et al.'s tree(s), because F. longipetiolata also exchanged genetic material at some point in the past with F. lucida after both species lineages were formed: they are reciprocally periphyletic. At the same time it homogenised its gene pool, which F. hayatae s.l. didn't (or couldn't: allopatric cryptic speciation) do. The only known precursor (FCA) of F. lucida (†F. altaensis ?) must have been closer to the common ancestor(s) of F. crenata + F. sylvatica-orientalis than to those of the ‘southern’ F. longipetiolata-hayatae s.l. Lineages leading to the latter two can only be addressed as monophyletic as their morphologies haven't evolved and lack (molecular-defined) lineage(s)-unique features. We could collect and classify them (and the modern-day F. longipetiolata) in a paraphyletic group of undiagnostic, ancestral morphologies: an explicit East Asian stem group (which the PhyloCode or APG would despise off, of course). Fagus hayatae s.l. comprises two cryptic (sister) species: the genetically rather primitive continental F. pashanica (longer ongoing gene flow with nearby beeches) and the insular, genetically clearly evolved F. hayatae s.str. (increased genetic drift). While the latter is clearly holophyletic (genetically highly coherent), its continental sister isn't; instead it is characterised by an ancestral genotypic polymorphism and could be paraphyletic. It's also possible that it mingled with other continental Asian species (including extinct ones) before (sorted out ...) or after (... never present in F. hayatae s.str.) the populations got separated from their insular sister (i.e. is epi- or periphyletic).
The North American species have a shared common origin, the North American lineage is monophyletic. But it remains to be seen if the two species (treated as subspecies in the Flora of North America) are reciprocally holophyletic. Genetically, F. mexicana – the today isolated Mexican populations – is closer to the common ancestor of the entire ‘Subgenus Fagus’ but also the ancestors of ‘Subgenus Engleriana’ (paraphyletic?), while F. grandifolia s.str. is generally well-sorted (holophyletic). A unknown variable are the extinct western North American beeches, which morphologically fit into the modern-day North American group but may or may have not had different genetic signatures. Their lineage extends to both sides of the Beringian Land Bridge. Interestingly, genotypes of F. grandifolia s.str. (in the north, hence, closer to the oldest known Paleocene and lower Eocene beeches) represent evolved (derived) variations of the ancestral genotypes still found in F. mexicana. The New World lineage to which both extant species belong (F. grandifolia s.l., i.e. according Flora of North America) is periphyletic because a sister lineage of modern-day F. grandifolia (s.str.) reached via North Atlantic Land Bridge into Europe, where their genetic characteristics were passed on into F. sylvatica. This seems to have happened after the North American gene pool started to split into a northern and southern gene pool.
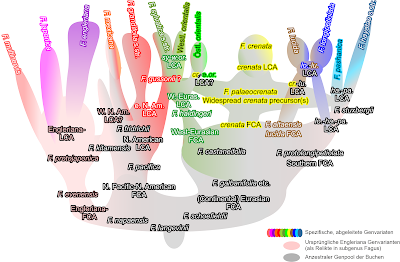 |
| The coral including the first (FCA) and last common ancestors (LCA) for orientation, and the main fossil taxa shuffled in (based on Denk & Grimm 2009; Renner et al. 2016, fig. 2). |
Interpreting the various reconstructions (tree and network inferences) towards the phyly of species, starting from a flexible empirical/ evolutionary species concept (cf. Mallet's papers) and explicitly including the fossil record (ancestor-descendant relationships; Denk & Grimm 2009; Renner et al. 2016), is pretty straightforward. Once we put the slavish (and boring) cladistic tree-thinking (no clade = "non-monophyletic") to rest and start exploring our wonderfully complex intra-generic data. And ask ourselves:
Further reading regarding our definition of species (including classics: no-one can say we didn't know 20 years ago):
Brown RM, Diesmos A. 2002. Application of lineage-based species concepts to oceanic island frog populations: The effects of differing taxonomic philosophies on the estimation of Philippines biodiversity. Silliman J. 42:133–162.
Cavender-Bares J, Ackerly DD, Baum DA, Bazzaz FA. 2004. Phylogenetic overdispersion in Floridian oak communities. Am. Nat. 163:823-843.
Funk DJ, Omland KE. 2003. Species-level paraphyly and polyphyly: Frequency, causes, and consequences, with insights from animal mitochondrial DNA. Ann. Rev. Ecol. Evol. Syst. 34:397-423.
Holland B, Huber KT, Moulton V, Lockhart PJ. 2004. Using consensus networks to visualize contradictory evidence for species phylogeny. Mol. Biol. Evol. 21:1459-1461.
Mallet J. 1995. A species definition for the Modern Synthesis. Trends Ecol. Evol. 10:294-299. [PDF]
Mallet J. 2001. The speciation revolution. J. Evol. Biol. 14:887-888.
Mallet J. 2007. Hybrid speciation. Nature 446:279-283. [PDF]
Mallet J. 2008. Hybridization, ecological races, and the nature of species: empirical evidence for the ease of speciation. Phil. Trans. Roy. Soc. B 363:2971–2986.
Mallet J. 2010. Why was Darwin’s view of species rejected by twentieth century biologists? Biol. Philos. 25:497-527.
Manen J-F. 2004. Are both sympatric species Ilex perado and Ilex canariensis secretly hybridizing? Indication from nuclear markers collected in Tenerife. BMC Evol. Biol. 4:46.
Muir G, Fleming CC, Schlötterer C. 2001. Three divergent rDNA clusters predate the species divergence in Quercus petraea (Matt.) Liebl. and Quercus robur L. Mol. Biol. Evol. 18:112-119.
Pikaard CS. 2001. Genomic change and gene silencing in polyploids. Trends Genet. 17:675-677.
Templeton AR, Robertson RJ, Brisson J, Strasburg J. 2001. Disrupting evolutionary processes: The effect of habitat fragmentation on collared lizards in the Missouri Ozarks. Proc. Natl Acad. Sci. USA 98:5426-5432.
Yatabe Y, Murakami N. 2003. Recognition of cryptic species in the Asplenium nidus complex using molecular data — a progress report. Telopea 10:487–496.
Cited literature
Ashlock PD. 1971. Monophyly and associated terms. Syst. Zool. 20:63–69.
Bomfleur B, Grimm GW, McLoughlin S. 2017. The fossil Osmundales (Royal Ferns)—a phylogenetic network analysis, revised taxonomy, and evolutionary classification of anatomically preserved trunks and rhizomes. PeerJ 5:e3433.—open access
Bryant D, Moulton V. 2004. Neighbor-Net: An agglomerative method for the construction of phylogenetic networks. Mol. Biol. Evol. 21:255-265.
Camus A. 1934. Quelques diagnoses de Fagacées [Diagnoses for Fagaceae]. Bull. Soc. bot. France 81:814–818.
Camus A. 1936–1954. Les Chênes : Monographie du genre Quercus [Monography of genus Quercus]. 3 vols. Paris: Paul Lechevalier.
Cardoni S, Piredda R, Denk T, Grimm GW, Papageorgiou AC, Schulze E-D, Scoppola A, Shanjani PS, Suyama Y, Tomaru N, Worth JRP, Simeone MC. 2021. 5S-IGS rDNA in wind-pollinated trees (Fagus L.) encapsulates 55 million years of reticulate evolution and hybrid origins of modern species. Plant J. doi:10.1101/2021.1102.1126.433057.—open access
Denk T. 1999. The taxonomy of Fagus in western Eurasia, 1: Fagus sylvatica subsp. orientalis (= F. orientalis). Feddes Repert. 110:177-200.
Denk T, Grimm GW, Hemleben V. 2005. Patterns of molecular and morphological differentiation in Fagus: implications for phylogeny. Am. J. Bot. 92:1006–1016.
Denk T, Grimm GW. 2009. The biogeographic history of beech trees. Rev. Palaeobot. Palynol. 158:83–100.
Denk T, Grimm GW. 2010. The oaks of western Eurasia: traditional classifications and evidence from two nuclear markers. Taxon 59:351–366.
Denk T, Grimm GW, Röseler A-K. 2012. When field botany meets history: taxonomy of Platanus mexicana in Mexico. Willdenowia 42:99–115.—open access
Grimm G. 2020 (updated 2021). Fagaceae collection. Dataset. figshare. https://doi.org/10.6084/m9.figshare.11603547.v3—open data
Grimm GW. 2003. Tracing the mode and speed of intrageneric evolution - a case study of genus Acer L. and Fagus L. Dr rer. nat. thesis Eberhard-Karls University.—open access
Grimm GW, Denk T, Hemleben V. 2007. Evolutionary history and systematic of Acer section Acer - a case study of low-level phylogenetics. Plant Syst. Evol. 267:215-253.
Grimm GW, Denk T. 2010. The reticulate origin of modern plane trees (Platanus, Platanaceae) - a nuclear marker puzzle. Taxon 59:134-147.—when you read this paper, read this one (written by a nigh-expert on “all -ics”): Nixon KC, Poole JM. 2003. Revision of the Mexican and Guatemalan species of Platanus (Platanaceae). Lundellia 6:103-137. And after that: Denk et al. (2012). Cladistics are not "all -ics", but a (too simple) philosophical subcategory of limited applicability at the species level and above.
Grimm GW, Denk T. 2014. The Colchic region as refuge for relict tree lineages: cryptic speciation in field maples. Turk. J. Bot. 38:1050–1066. [PDF]
Jiang L, Bao Q, He W, Fan D-M, Cheng S-M, López-Pujol J, Chung MG, Sakaguchi S, Sánchez-González A, Gedik A, Li D-Z, Kou Y-X, Zhang Z-Y. 2021. Phylogeny and biogeography of Fagus (Fagaceae) based on 28 nuclear single/low-copy loci. J. Syst. Evol. doi:10.1111/jse.12695.
Lepoittevin C, Bodénès C, Chancerel E, Villate L, Lang T, Lesur I, Boury C, Ehrenmann F, Zelenica D, Boland A, Besse C, Garnier-Géré P, Plomion C, Kremer A. 2015. Single-nucleotide polymorphism discovery and validation in high density SNP array for genetic analysis in European white oaks. Mol. Ecol. Resources 15:1446–1459.
Liede-Schumann S, Meve U, Grimm GW. 2019. New species in Drosanthemum (Aizoaceae: Ruschioideae). Bradleya 37:226–239.
Mayr E, Bock WJ. 2002. Classifications and other ordering systems. J. Zool. Syst. Evol. Res. 40:169-194.
Neophytou C, Aravanopoulos FA, Fink S, Dounavi A. 2010. Detecting interspecific and geographic differentiation pattern in two interfertile oak species (Quercus petraea (Matt.) Liebl. and Quercus robur L.) For. Ecol. Managem. 259:2026–2035.
Pojárkova AI. 1933. Botanico-geographical survey of the maples of the USSR in connection with the history of the whole genus Acer L. Acta Inst. Bot. Acad. Sci. USSR, Ser. 1 1:225-374. — I only had a paper copy of the paper; which are all gone (I didnt't move paper) and so far failed to find the original online.
Renner SS, Grimm GW, Kapli P, Denk T. 2016. Species relationships and divergence times in beeches: New insights from the inclusion of 53 young and old fossils in a birth-death clock model. Phil. Trans. Roy. Soc. B 371:20150135.
Schwarz O. 1936. Entwurf zu einem natürlichen System der Cupuliferen und der Gattung Quercus L. [Sketch for a natural (i.e. phylogenetic) classification system of cupulifera and oaks] Notizbl. Bot. Garten Museum, Berlin-Dahlem Vol. 13 (Nr 116):1–22.
No comments:
Post a Comment
Enter your comment ...