I always have had the impression that genetic papers are rarely reviewed by people with any idea about the studied organism. Traditionally, this was of little concern. Genetics focussed on processes not phylogenetics. But, with the advent of next-generation sequencing techniques, anyone can now tap into large data sets. Thanks to the advance in computer programming and the effort developers put in tutorials and help pages (e.g. BEAST, IQTree, RAxML, dedicated GoogleGroup), one doesn't need any particular knowledge or prior experience to analyse the data. Phylogenomic datasets become more and more common, and we have more and more complete plastomes to play with.
Although oaks are the largest extratropical tree genus – an oak was among the first trees for which a full plastome was generated and annotated, assembled at a time when it still required a lot of different skills and resources – they have long been ignored and, until very recently (Zhou et al. 2021 adding a good and well-sampled bunch), we had relatively few (published) complete plastomes (there are a score of unpublished ones, I'll come back to this later). And even though all chromosomes of the most important western Eurasian oak, Quercus robur – introduced as shipwood or ornamental tree across the globe by the British – have been sequenced, you couldn't find a complete plastome of that species in gene banks.
The P. R. China has not only a lot of research money but also a huge scientific workforce pool, hence, it's no surprise that Chinese groups now start sequencing one overlooked genus after the other, and so we finally have more than one complete oak plastome.
And have enriched scientific literature with now dozens of completely useless phylogenetic trees and phylogeny-related inferences, statements and conclusions (but see Zhang et al. 2020, for a notable exception, already the title is a refreshing detour from the dead-beaten path of plastome-based phylogenetics; see also Why you never should do a single-species plastid analysis of oaks).
An updated classification of oaks
Until very recently, researchers had to rely on outdated systematic systems, some of which were in not too bad fit with molecular data accumulated in the last 20 years. However, none got it right.
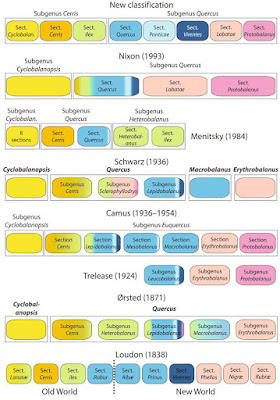 |
| Oak classifications through time (Denk et al. 2017, fig. 2.1). Colours represent main intra-generic phylogenetic lineages (species groups recognisable by coherent nuclear-genetic and morphological differentiation patterns) |
The latest addition was the system by Nixon (1993). Kevin C. Nixon is the "de-facto expert on North American oaks" (self-description), a faithful follower of Steve Farris, the cladistic demi-god. He obviously has a lot of knowledge about oaks (hence: Nixon 1997), and can be credited for describing new species in the Americas. Intriguingly, the cladistic analysis cited in the 1993 paper never was published. De-facto expert Nixon, who invented and coded a fast parsimony implementation (Nixon 1999), seems to have only co-authored a single molecular phylogenetic paper on oaks (Manos et al. 1999). Probably incomprehensible to him, being an renown expert on "all -ics", already this first molecular data – a classic region: the ITS regions of the 35S nuclear-ribosomal(nr)DNA cistron – rejected his systematic views, like pretty much any other molecular data that covered his other morphological-cladistic works (e.g. in case of plane tree, Platanus: Nixon & Poole 2003, cf. Grimm & Denk 2008, 2010; Denk et al. 2012; De Castro et al. 2013). A man of principle, he just keeps on ignoring the molecular evidence proving him wrong. Being prominent in (palaeo-)botanical circles, he can do so, as recently demonstrated by Science (Wilf et al. 2019; Surviving parsimonists: just tree-naive or tree-blindfolded?)
In 2009, we studied the oak pollen and found a very good fit with earlier (nuclear-data) based molecular phylogenies (Manos et al. 2001) and established a system that recognised six informal groups (Denk & Grimm 2009).
The paper got published despite fierce fire from an "anonymous" expert, suspectedly William L. Crepet (old pal of Nixon's: e.g. Crepet & Nixon 1989, a classic piece demonstrating how they and their disciples still work 30 years and a few revolutions later, e.g. Wilf et al. 2019). This reviewer's main critique was, “I know this since 25 years but I never found it worth publishing”. Well, we did. With more and more molecular data accumulating and enforcing what we found, we finally ceased the occassion to formalise our groups in a introductory book section (Denk et al. 2017; reviewed by taxonomists familiar with the workings of the Botanical Code).
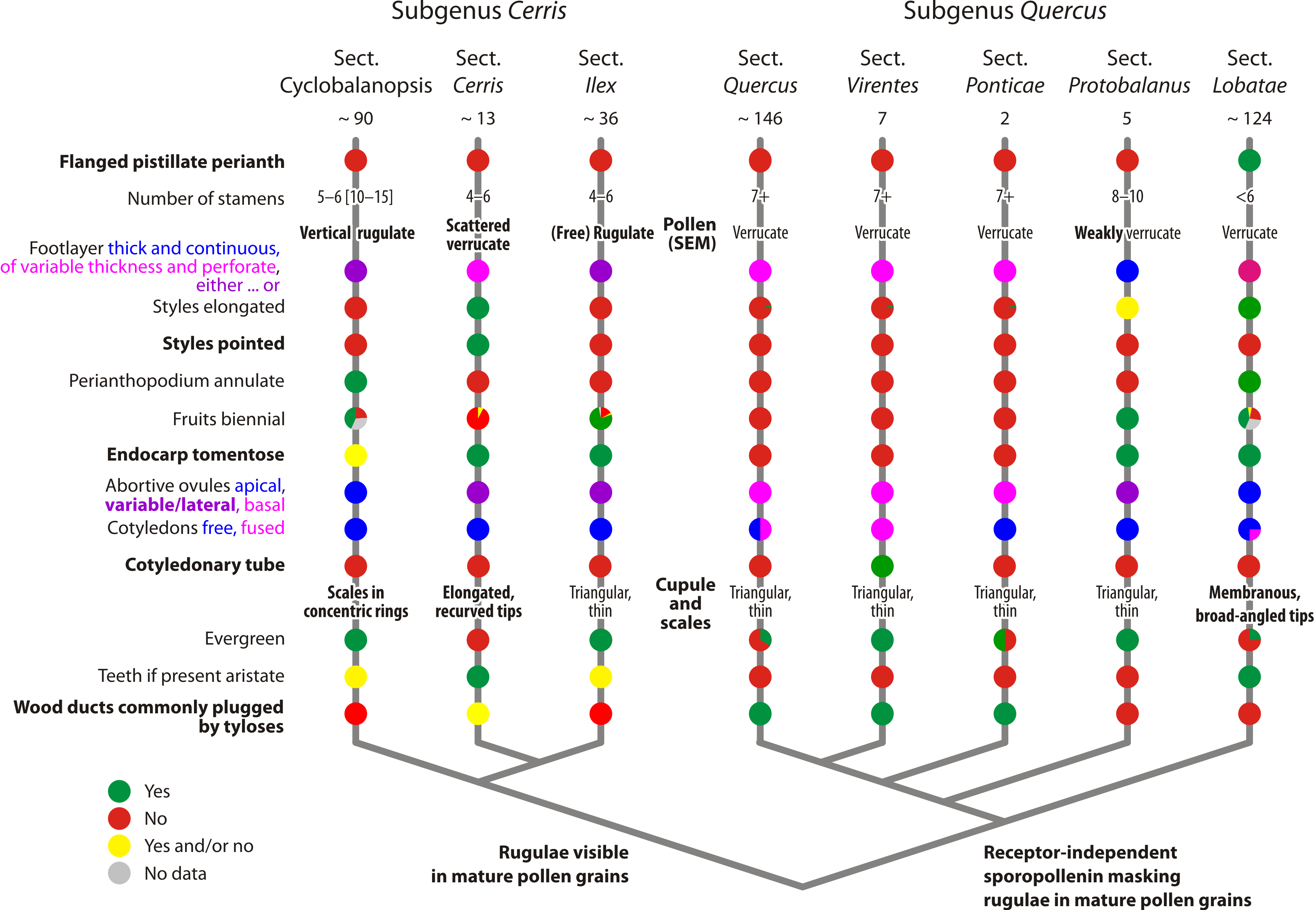 |
| A nuclear-data based systematic framework for oaks (genus Quercus). |
Knowing that the book chapter will possibly be lost, we published the pre-print, accessible for all, on bioRxiv, and providing a species table and other supplementary information in an open-data spread-sheet via figshare.
In the beginning only few picked it up. In most oak-related literature (being not reviewed by anyone still working systematically or phylogenetically on oaks), the authors sticked to outdated systems or local traditions. This has apparently changed.
What we know: nuclear data 'Yay', plastid data 'Nay'
Oaks fall into two main evolutionary lineages, which we formalised as subgenera.
The mostly New World (Americas) subgenus Quercus is opposed by the exclusively Old World (Eurasian) subgenus Cerris.
Phylogenetic relationships within subgenus Cerris are resolved since Hipp et al. (2019; see also Zhou et al. 2021, fig. 2): section Cyclobalanopsis represents the first diverging branch, followed by the, reciprocally monophyletic (today holophyletic, cf. Ashlock 1971), sister sections Cerris and Ilex. At some point in the past, section Ilex might have been paraphyletic, with the first Cerris evolving from an Ilex-like ancestor.
Within subgenus Quercus, we are still a bit uncertain about the sequence, but it seems that section Lobatae is the first diverging branch, followed by section Protobalanus, section Ponticae, section Virentes and finally section Quercus. The data is conclusive that all sections are monophyletic in a general sense. The species-poor modern-day Protobalanus, Pontica, and Virentes are likely holophyletic. Being a relict, less-evolved lineage, section Virentes can inflict topological ambiguity pending on the sect. Quercus sample used to infer phylogenetic trees.
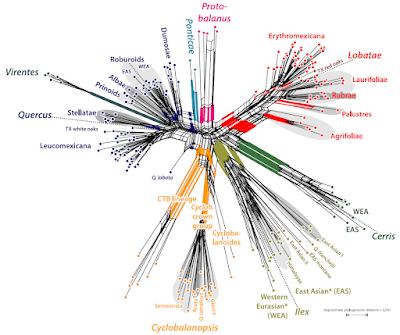 |
| A SNP distance-based meta-phylogenetic network of oaks (Hipp et al. 2019, fig. 6). Not familiar with such graphs? See Next-generation neighbor-nets for a walk-through) |
And we know the sections are old, they root deep.
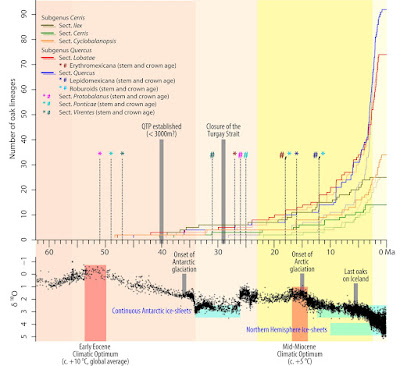 |
| A lineage-through-time plot for oaks (after Hipp et al. 2019, fig. 2) |
We also know (for quite some time) that even though the sections are monophyletic, mostly holophyletic, they do exchange genetic material from time to time (within their respective subgenera), and did so also in the past. This, in addition to the often very large active population sizes and the general permeability of species boundaries within the sections, messed up a lot: traditional nuclear markers suggested as species barcodes such as the ITS1 and ITS2 of the 35S rDNA can be a pain, with higher intra-genomic than inter-species divergence (classic papers: Samuel et al. 1998; Muir et al. 2000, 2001; see Denk & Grimm 2010 for the broadest-sampled nrDNA spacer data so far; and Piredda et al. 2020 for a small-sample deep dive); and plastids simply ignore species boundaries. In general, nevertheless, the nuclear signatures fit with the species determinations and trees (or networks) based on nuclear data make a lot of sense regarding morphology, ecology and history. For instance, a section Ilex oak may have quite different ITS copies (some shared between different species), but all of them are unique to section Ilex. The plastid signatures, on the other hand, can be utterly at odds.
A classical case is that of Q. alnifolia (endemic) and Q. coccifera (widespread in the Mediterranean), two Ilex oaks species sharing the island of Cyprus. They are sympatric but morphologically distinct. The nuclear data had a close to 100% match with the morphology. The plastid data also revealed two distinct haplotypes, which, however, seemed to be randomly distributed among individuals of both species (Neophytou et al. 2010, 2011).
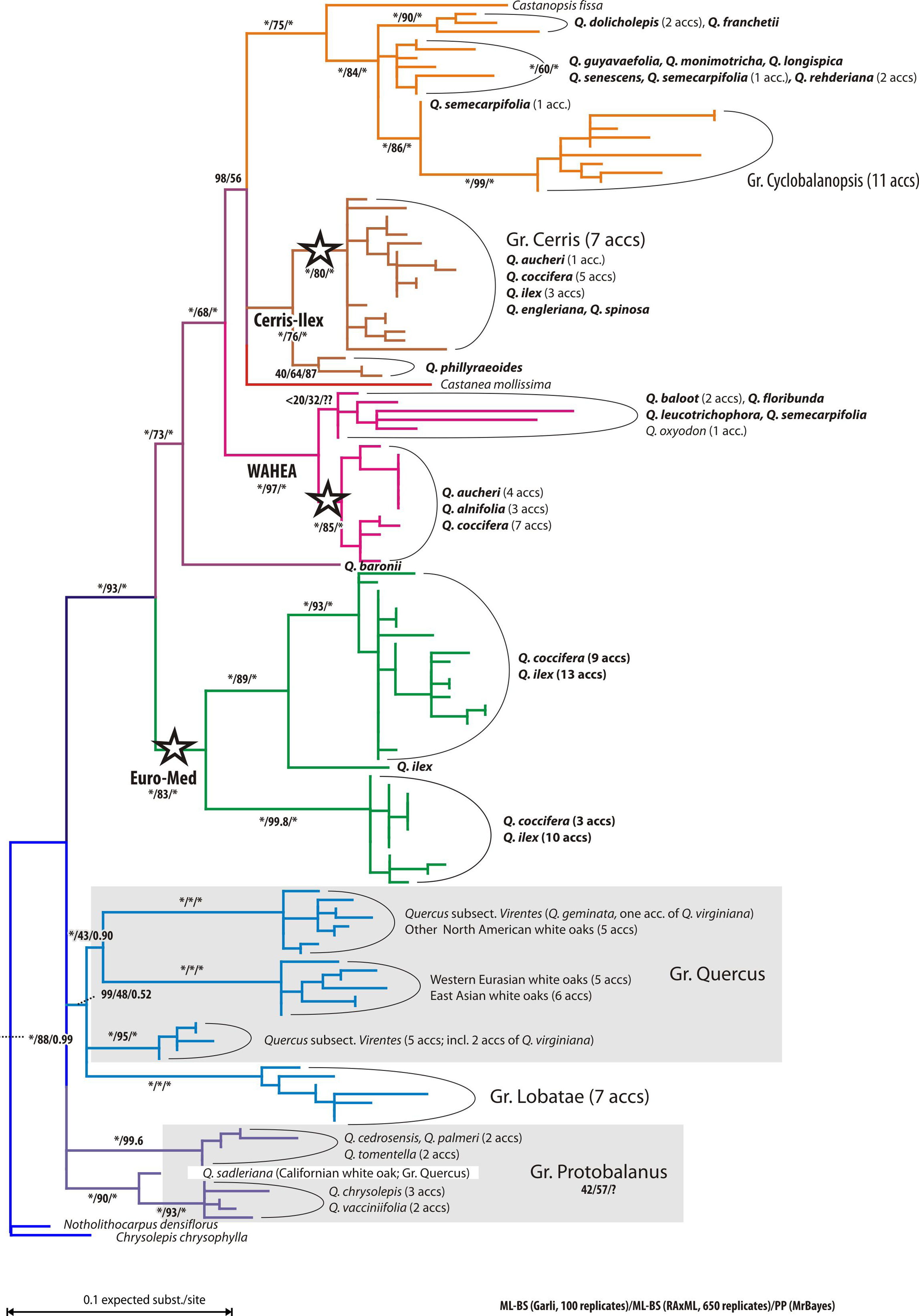 |
| A “fragment” plastid phylogeny focussing on western Eurasian accessions of sections Ilex (Simeone et al. 2016, fig. 1). Tree is rooted with sequences of two (western) North American relict Fagaceae genera. Note the seemingly para-/polyphyly (“non-monophyly” in contemporary literature) of Q. ilex plastids (stars), engulfing East Asian siblings as well as section Cylcobalanopsis and accessions of other Eurasian Fagaceae: the chestnuts (Castanea) and their (sub)tropical sister genus Castanopsis. |
The western Eurasian members of sections Cerris and Ilex share a haplotype (‘Cerris-Ilex’), and while Cerris seems to be homogenous, Ilex oaks allow themselves the luxury of two more, visibly different haplotypes of different phylogenetic affinity (‘WAHEA’ and ‘Euro-Med’). In East Asia, haplotypes of the same lineage can be found in sections Ilex and Cyclobalanopsis (yellow clade). North America has not been studied a lot regarding plastome differentiation until Zhou et al. (2021), but the unpublished 80+ complete plastomes I happen to get in contact with five years ago, were unambiguous: it matters less whether you are a red (sect. Lobatae) or white oak (sects Ponticae, Quercus, Virentes; very different ITS sequences and phylogenomic SNP patterns). But it matters a lot where you come from (western North America or eastern North America, New World or Old World). Which is probably one reason, they are still unpublished. What can you do with a phylogenomic data set that misses out on the most principal phylogenetic relationships?
And it doesn't stop at the genus level. Way back in 2003, Cannon & Manos noted a shared haplotype in Castanopsis and Lithocarpus (again, trivial to distinguish based on ITS data). In 2008, Manos et al. published an undercited paper on Notholithocarpus, a newly recognised Fagaceae genus, in a regional low-impact journal. In that publication one can find the only sensible Fagaceae oligo-gene tree for a decade to follow, based on a combined nuclear-plastid data set (all other trees based on mixed or plastid-only data, e.g. according subtrees in Li et al. 2004, Sauquet et al. 2014, Xing et al. 2014, Xiang et al. 2014, Larsson 2016 are for the bin).
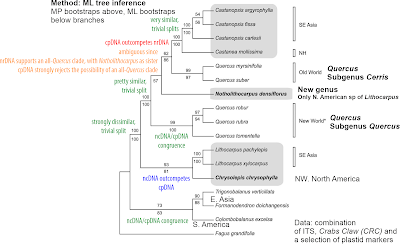 |
| An early oligogene tree combining nuclear (nc) and plastid (cp) sequence data (after Manos et al. 2008). The (today) holophyletic oaks are “not monophyletic”, with the New World oaks drawn away and the Old World oaks drawn to the Eurasian Castaneoideae (a paraphyletic subfamily). The geographic attraction is only disrupted in the deeper part, where the western North American temperate Chyrsolepis (originally treated as a Castanopsis) comes as sister of the (sub)tropical stone nuts (Lithocarpus). |
Which brings us to the recently published sino-centric complete plastome phylogenies.
When the plastome isn't species-sorted, how can it inform phylogenies?
A first honest complete plastome Fagaceae tree can be found in fig. 2 of Worth et al., PeerJ (2019).
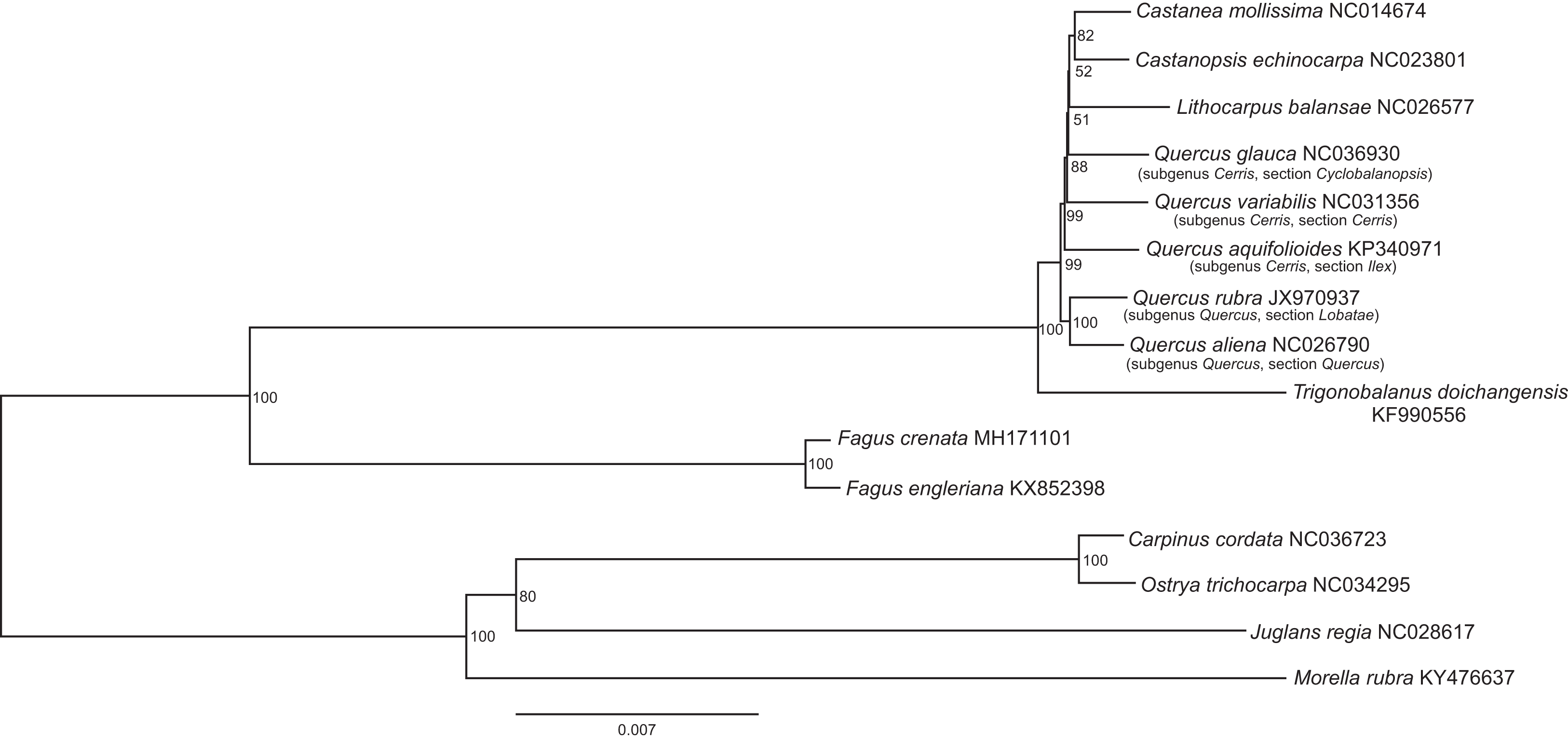 |
| A tiny tree based on complete plastomes that shows a lot. |
Worth et al.'s focus (they also published a pre-print) was introducing a second complete Fagus plastome, Fagus crenata, and the group has since assembled a nice and extremely puzzling complete plastome sample (Worth et al., 2021). Fagus is an equally overlooked but highly interesting Fagaceae genus which includes a familiar tree, the ‘Common Beech’. What makes Worth et al.'s tree particular compared to most other complete plastome trees, is the use of branch-lengths: Obviously Fagus, the beech, has little to do with the rest of the family, the Fagaceae. And within the Fagaceae, most of the complete plastome variations is tip-limited, while the deeper branches are well supported but nearly non-existant. The outgroups, other Fagales, cannot possibly provide any useful information, being lightyears (more exactly > 80 million years) away. The overall topology in this few-tip tree is nonetheless very typical: the without a doubt monophyletic-holophyletic oaks – Nature dices a lot, but you don't get exactly the same suite of characters by chance – always get separated into mostly New World (subgenus Quercus) and Old World (subgenus Cerris) oaks forming reciprocal clades.
 |
| Two recently published “phylogenetic results” in the form of ML-inference based cladograms making use of complete plastome sequences of oaks (and other Fagaceae, upper left). Main aspects – "non-monophyly" of Quercus and sect. Ilex – and errors – commonly found in similar papers – annotated. Note that both trees were published in non-predatory, officially peer-reviewed, scientifically edited journals (Mitochondrial DNA Part B as “Mitogenome[sic!] Announcement” and Wiley's Ecology & Evolution) |
And in the latter's clade, we find the (today) Eurasian Castaneoideae (Lithocarpus, Castanea + Castanopsis) deeply nested. Which is puzzling, since according to flagship journal Science and Nixon's “cladistic analysis”, Castanopsis originated in the Eocene of Patagonia and migrated (without leaving a tangible trace, not even a single pollen grain) via Antarctica and Australia into S.E. Asia; apparently it lost its plastid on the way and took those of the (always) Eurasian chestnuts after pushing oaks and stone nuts aside.
Although I find it very laudable that complete plastomes are generated for oaks (or other Fagaceae), one should be very careful with the relevance of complete plastome papers like the two above for oak phylogeny and (genetic) taxonomy (see also my comment to Yang et al. 2018, Frontiers in Plant Science, showing the same deficits). Or biogeography, when all samples comes from the territory of the P.R.C. (ideally including Taiwan, for bonus points with the local stakeholders).
... our findings do not support previous research which retrieve Quercus subg. Cerris sect. Ilex as a monophyletic group, with sect. Ilex found to be polyphyletic [which means: even though their nucleomes and morphology are coherent, they don't share a common ancestry; i.e. it's all random similarity due to convergent evolution] and composed of three strongly supported lineages inserted between sections Cerris and Cyclobalanposis. — copied from the abstract of Li et al. (2021).
It's pretty revealing for the scrutiny of the review process, when Chinese authors don't even spell their native taxa right ("...anpos..." instead " ...anops...", a classic misspelling error of Chinese-speakers; remembering the sign at the highway near Pinyin: "Museum of Natural Hisorty")
Persistently done in complete plastome (phylogenomic) studies, one should not ignore what broadly sampled plastid markers have shown and is long known:
Taxonomy, speciation, is largely decoupled from plastome differentiation in oaks and other Fagaceae.
Hence, a few or even many complete plastomes cannot be used to put forward any phylogenetic hypothesis for species-rich genera like oaks. Or provide a phylogenetic (or "cladistic", as de-facto expert Nixon would call it) test for our updated or any other systematic framework. That a species or genus doesn't form a high-supported clade in a chloroplast tree doesn't provide any reason to reject holophyly (or even monophyly in a general sense). When we put our's up, we based it exclusively on nuclear data because we knew for over a decade that the plastids are not sorted during speciation. Especially not, if samples are from cultivars of unknown provenance and from the same geographic area. And a genus phylogeny is tasked to represent a species tree. One needs data reflecting speciation events.
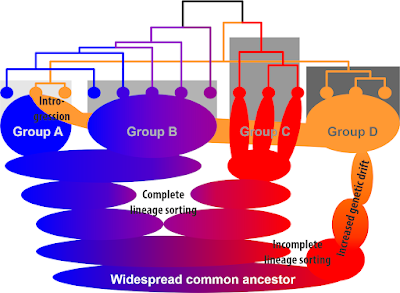 |
| A simple scenario demonstrating how easy it is for plastomes to decouple from phylogeny. The speciation processes (isolation of gene pools, bubbles) only sort the plastomes to some degree, secondary re-radiations will lead to plastomes being captured from ± distant sibling lineages. The potentially inferred (ultrametric) tree (top) cannot possibly depict the phylogenetic relationships between sampled species (or populations). In a situation like this, only two of the holophyletic groups would be resolved as clades, one (group D) including morphologically non-fitting specimens (intrograded members of group A). The sorting in the most widespread, most diverse group B will lead to a grade because its plastid haplotypes show higher or lower sequential similarity to haplotypes of group A vs. group C. |
Or for Fagaceae in general, as now finally demonstrated by Zhou et al. (2021): Fagaceae plastids are generally a phylogenetic mess informing us about
- past reticulations, hybridisation, introgression and complete take-overs—by finding a wrong or too divergent plastomes in a nuclear-morphological coherent group such as e.g. modern-day section sect. Ilex;
- but not the evolutionary history, the species tree—probably a species network in the case of oaks and other Fagaceae/ Fagales (see Cardoni et al. 2021, and literature cited therein)
It also cannot be used to test phyly of their taxa (see also: Monophyletic species). Long ignored – despite producing the most interesting signals in Manos et al.'s 2008 data set – Zhou et al. included complete plastomes of the relict genera Formanodendron (China), Colombobalanus (South America), Notholithocarpus and Chrysolepis (western North America). And they confirmed what was already apparent from the 2008 data and any plastid "fragment" sequenced in the last 20 years: the chloroplast genomes are geographically but not phylogenetically sorted, even above the genus level!
 |
| Zhou et al. (2021), fig. 2: Left, the Fagaceae species tree based on 2000+ nuclear gene regions; right, the systematically largely decoupled plastome genealogy (complete chloroplast genomes). The three main East Asian sect. Ilex haplotypes annotated in pink; red dots indicate the likely placements of the not sampled (naturally, hard to get in China or the U.S.) western Eurasian (Hindukush to Atlantic) haplotypes of section Ilex (cf. Simeone et al. 2016). |
I'm curious, when this realisation reaches also the last editors and reviewers of the many (allegedly) peer-reviewed journals. There's quite an illustrous list of data-/inference-naive publications on oaks, Fagaceae and Fagales, including even high-fly journals such as Systematic Biology in 2012 (only Nixon's wife and his disciple Wilf are co-authors; one wonders who anonymously scrutinised that paper without any conflict of interest) and New Phytologist in 2016 (single-author, Nixon and other U.S. big shots acknowledged). Willful less-impact venues are Frontiers in Plant Science (often P.R.C.-edited and -reviewed papers), Journal of Systematics and Evolution (P.R.C.-owned), and Mitochondrial DNA B, the last standing journal that will publish a single new chloroplast genome sequence as "Mitogenome Announcement" and no matter what tree. Any new plastome is new data, filling a white spot, hence, worth publishing in some form (material should be georeferenced, though, so others can make use of it). But if you have no idea how to infer a meaningful tree, just don't! No tree is always better than a poorly done one.
What hold for oaks and Fagaceae, holds also for any other plant where plastids are inherited from the mothers only.
If species of the same (nuclear-unambiguous) lineage or widespread species lack lineage-diagnostic, sorted plastid signatures, any plastid data, partial or complete chloroplast genomes, are useless for taxonomy and systematics.
And unless linked to same-individual nuclear data, plastid data can be problematic for phylogenetics in general.
Especially, when one wants to stick to a cladistic classification—which I, working evolution's coalface, naturally don't give a shit about (What is an angiosperm? [intro][pt 1][pt 2]; Clade, cladograms, cladistics, and why networks are inevitable), but relevant people still do such as the Angiosperm Phylogeny Group (APG) and about 4 out of 5 reviewers that judged our papers. When your plastid genealogy is in strong conflict with morphology, it's probably not well sorted during speciation and phylogenetically misleading. Nuclear data is then without alternative.
What can complete oak plastomes provide
Letting aside taxonomy and phylogenetic (or trivial cladistic) systematics, complete plastomes could be highly useful.
Their main information content at this point is to find new marker regions for large-scale haplotype studies at various hierachical levels. To enable us to map how incongruent plastomes are with their vectors, the populations and (various) species carrying them. Pin-point when plastomes were sorted by geographic (climate-triggered) vicariances and dispersals and speciation events, and when not. So far, we effectively only have the trnH-psbA spacer to study intra- and interspecific geographic differentiation. All the other traditionally used plastid "DNA barcodes" (used in the above cited all-Fagales studies) lack the divergence to bring light into the many dark spots below genera.
As done by Zhou et al. (2021) for the family, a future application will be to pinpoint the decoupling of plastid geneaologies from the species phylogeny, the nuclear-morphological evolution, in space and time. Since the main oak lineages have been around for tenths of millions of years, we need to cover ground, geographic space, not taxonomic space (species).
So far, however, all complete chloroplast genome studies – focussing on Chinese microspecies – have revealed little beyond what we already knew from “DNA fragments” (used as a prerogatory term in complete plastome studies), i.e. individual markers. Only by using a few of those “fragments” but a global sample, Pham et al. (2017) and Yan et al. (2019) could extract much more about the history of oaks, their past migration patterns and what triggered them than any complete plastome study. Since plastid signatures are geographically highly constrained, they can tell us where a population and its (maternal) ancestors are from, even if it's in the wrong species. Species don't capture plastomes from thousand of kilometers afar, they get them from those species that, at the time, thrived nearby. If two contemporary species share a plastome but are not sympatric, we directly know their ancestors must have been. The more basepairs, the easier it is for the algorithms to estimate rate changes, and the better a dating. Zhou et al. (2021) represents a first step, but it's only a (data-wise massive but still) scratch of the berg lying under the ice. It's not hard to imagine what evolutionary treasures, we could dig out about the dynamic geographic history of oaks, if we would have complete plastomes from the critical provenances all over the world. For instance, Zhou et al. (2021) didn't sample the most-distinct "WestMed" haplotype lineage of Q. ilex or included any western Eurasian spp. of section Ilex (cf. Simeone et al. 2016; Vitelli et al. 2017). Critical East Asian species of section Ilex such as Q. baronii and Q. phillyreoides (bridging between Ilex- and Cerris-oaks, morphologically and/or genetically) or the Himalayan Q. semecarpifolia, physically bridging between the two centres of Eurasian oaks, are missing, too (cf. Pham et al. 2017, Yan et al. 2019). They also only scratched the surface of the New World oaks. There's still a lot to research on Fagaceae/ Fagales in general, and oaks in particular.
Until then, the beeches (Worth et al., 2021; The challenging and puzzling ordinary beech – a [hi]story; A fully resolved, and perfectly misleading, species tree) will become the blueprint for what may still be under the surface of Fagaceae/ Fagales. So, stay tuned. And Happy Christmas everyone.
Cited literature
No comments:
Post a Comment
Enter your comment ...